阿尔茨海默病(Alzheimer's disease,AD)是发生于老年和老年前期、以进行性认知功能障碍和行为损害为特征的中枢神经系统退行性病变。临床上表现为记忆障碍、失语、失用、失认、视空间能力损害、抽象思维和计算力损害、人格和行为改变等。
近年来研究显示,突触(树突棘)丢失是导致AD认知障碍的主要原因。Cdc42是小G蛋白Rho亚家族的成员,在调节突触可塑性中起着重要作用。Cdc42GAP是Cdc42活性的负调控因子,调控其从活性状态转化为非活性状态。不过,目前很少有研究关注Cdc42在阿尔茨海默病进展中的作用。
近日,来自南方医科大学基础医学院的张璐和张琳教授团队发表了题为“Cdc42GAP deficiency contributes to the Alzheimer’s disease phenotype”的文章,展示了课题组最新的研究成果。该研究旨在探索Cdc42信号通路的激活是否与阿尔茨海默病样表型有关。
Cdc42GAP缺乏损害认知和空间记忆
研究者收集了11个月大的野生型(WT)小鼠、GAP(Cdc42持续激活小鼠,即Cdc42GAP敲除杂合小鼠)和AD模型小鼠(APP/PS1)的海马体和皮层组织,以检测Cdc42信号通路。结果显示,GAP小鼠和AD小鼠的海马体和皮层中活化的Cdc42均显著增加;且Cdc42信号通路下游PAK和cofilin的磷酸化水平均有所升高,表明Cdc42GAP缺陷小鼠和AD小鼠中Cdc42的信号传导通路均发生活化。
此外,研究者将GAP小鼠与AD小鼠杂交,创建了同时具有AD小鼠表型和Cdc42GAP缺陷的GAP/AD小鼠,并使用4和11个月大的小鼠(分别代表年轻和老年阶段),探讨Cdc42GAP缺乏对学习及认知功能的影响。在新物体识别测试中,4月龄GAP小鼠已经出现明显的认知损伤,且GAP/AD小鼠认知功能损伤进一步加重,表现为与新物体互动时间更少,互动频率也更低(图1B-C)。而且,老年GAP小鼠表现出比年轻小鼠更严重的识别和记忆障碍。这些结果表明,Cdc42GAP缺乏会导致认知功能障碍,这种障碍可能会随着年龄的增长而恶化。
在避暗实验中,也发现与上述新体物识别测试类似的结果,表明Cdc42GAP缺乏与识别和记忆功能障碍也有关系。在Morris水迷宫实验中,GAP和GAP/AD小鼠显示出穿越目标象限平台的次数减少,将目标平台转换位置后,再次进行训练和检测,与上述结果相似(图1H-I),年长的GAP小鼠的表现比年轻GAP小鼠差,表明Cdc42GAP缺乏可能会损害空间记忆功能。
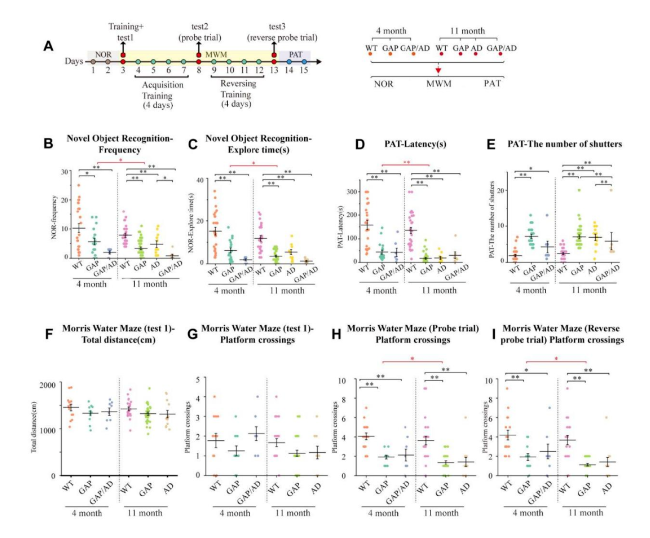
图1 Cdc42GAP缺乏导致认知异常,随着年龄的增长,出现更严重的行为缺陷
Cdc42GAP缺乏诱导神经元衰老和树突棘丢失并伴有F-肌动蛋白解聚
研究者以神经元衰老标志物SA-β-gal评估Cdc42GAP缺乏在神经元衰老中的作用。结果显示,年轻GAP小鼠的海马和皮层神经元中SA-β-gal水平显著升高,GAP/AD小鼠衰老更显著,而且年老GAP/AD小鼠的衰老程度明显更严重,表明Cdc42GAP缺乏诱导并加速了神经元衰老(图2)。
此外,研究者探索了Cdc42GAP在视觉皮层和海马两个脑区神经元树突棘可塑性中的作用。结果发现,Cdc42GAP缺乏会导致小鼠海马和皮层突触可塑性的长期损伤。通过检测年老小鼠海马和皮层中神经元树突棘的F-肌动蛋白和G-肌动蛋白,发现GAP和AD小鼠的海马和皮层中F-/G-肌动蛋白的比例显著降低,表明Cdc42GAP缺乏诱导树突棘丢失和F-肌动蛋白解聚。

图2 Cdc42GAP缺乏诱导神经元衰老
Cdc42GAP缺乏诱导并加速tau磷酸化和淀粉样蛋白β的产生
接下来,研究者使用年老小鼠研究Cdc42GAP缺乏在tau磷酸化和Aβ产生中的作用。免疫荧光分析显示,GAP小鼠的海马和皮层中的tau磷酸化(p-T231/AT8)水平显著升高(图3)。与GAP和AD小鼠相比,GAP/AD小鼠中的tau过度磷酸化进一步增加,表明Cdc42GAP缺乏可能有助于tau磷酸化的进展。另外,GAP/AD小鼠的Aβ1-42和Aβ1-40水平显著升高。这些结果表明,Cdc42GAP缺乏诱导并加速tau磷酸化和Aβ1-40和Aβ1-42的产生。

图3 Cdc42GAP缺乏在11个月大的小鼠中诱导阿尔茨海默病表型
Cdc42GAP缺乏可能通过激活GSK-3β诱导阿尔茨海默病表型
证明了LH Nts神经元的Nts和GABA共同释放介导了这种行为效应[Fig.4e-h]。为探索Cdc42GAP缺陷小鼠阿尔茨海默病表型的潜在机制,研究者对年老WT和GAP小鼠的海马进行了定量磷酸化蛋白质组学分析。结果发现,GAP小鼠微管相关蛋白tau(MAPT)许多位点发生磷酸化;同时,GSK-3βSer-389位点磷酸化水平显著下降。
接下来,研究者通过蛋白质印迹和免疫荧光验证发现GAP、AD和GAP/AD小鼠的海马和皮层中GSK-3βSer-9位点和Ser389位点磷酸化水平显著降低,而且GAP/AD小鼠GSK-3βSer-9和Ser389的磷酸化水平低于AD小鼠。此外,GAP、AD和GAP/AD小鼠的海马和皮层中磷酸化GSK-3βTyr-216的水平增加,其中GAP/AD鼠皮层中的Tyr-216水平高于GAP和AD鼠。
这些结果表明Cdc42GAP缺乏通过Ser-9、Ser-389处去磷酸化和/或在Tyr-216处磷酸化来激活GSK-3β,从而诱导并加速海马和皮层中的阿尔茨海默病样表型。
Cdc42持续激活诱导tau过度磷酸化和突触丢失
为验证Cdc42GAP缺乏在tau过度磷酸化和树突棘丢失中的机制,研究者制备了Cdc42组成型激活和显性负效突变慢病毒,转染进各组小鼠海马和皮质原代神经元,结果显示,与对照组相比,Cdc42持续激活诱导海马和皮质神经元tau磷酸化水平增加,而降低Cdc42活性诱导其tau磷酸化水平显著降低,表明Cdc42 GTP酶活性升高可能是阿尔茨海默病tau过度磷酸化的原因。
接下来,研究者通过分析小鼠海马和皮层原代神经元突触可塑性发现,Cdc42持续激活诱导海马和皮层神经元树突棘丢失和衰老水平升高,表明Cdc42 GTPase活性升高与阿尔茨海默病样树突棘丢失有关。
阿尔茨海默病患者大脑皮质切片中Cdc42信号通路和GSK-3β被激活
此外,研究者收集了AD患者和健康对照(HC)大脑的皮质石蜡切片标本,并通过免疫化学染色评估Cdc42信号通路和GSK-3β活性。结果发现,AD患者皮层中的Cdc42-GTPase与HC相比显著增加,磷酸化-cofilin(p-cofilin(Ser-3))和磷酸化-PAK1(p-PAK1(Ser-199))水平也有所增加,表明激活的Cdc42信号通路可能导致人类阿尔茨海默病相关神经元退行性改变(图4)。
此外,Aβ1-40、Aβ1-42和APP代谢产物CTFs在AD患者中显著升高,表明Cdc42GAP缺乏活化了Cdc42信号通路,激活的GSK-3β在阿尔茨海默病患者中起着至关重要的作用。

图4 阿尔茨海默病患者的皮层中Cdc42通路和GSK-3β活性被激活
原文链接:
https://doi.org/10.1093/brain/awad184
参考文献:
Zhu M, Xiao B, Xue T, et al. Cdc42GAP deficiency contributes to the Alzheimer's disease phenotype [published online ahead of print, 2023 May 31]. Brain. 2023;awad184. doi:10.1093/brain/awad184
产品订购:sales@amyjet.com
邮政编码:430070
公司地址:武汉市洪山区光谷大道35号
光谷总部国际二期时代1栋13楼
提示:本公司所有产品仅供科研使用,不用于临床诊断。
版权所有:艾美捷科技有限公司 鄂ICP备10204150号-1 鄂公网安备:42018502004523号
第二类医疗器械经营备案凭证:鄂汉药监械经营备20234324号

微信扫码在线客服